

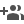







Researchers have decoded the molecular mechanism in which a cell surface receptor belonging to the family of enzymes that bind growth factors, regulate cell differentiation, proliferation, survival, metabolism, and migration, prevents cancers.
This enzyme called VEGFR1 withholds self-expression (autoinhibited) in the absence of a ligand—for example hormones. The research can show the way for developing medical solutions for colon and renal cancers by using molecules that preferentially stabilises the inactive state of VEGFR1.
Cell surface receptors like Receptor Tyrosine Kinases (RTK) are crucial for converting extracellular signals (from chemical cues like growth factors, generally referred to as ligands) to tightly regulated cellular response. Ligand binding to extracellular receptors activates intracellular coupled enzymes (tyrosine kinases). The activated enzyme, in turn, adds phosphate group to several tyrosine molecules that function as an adaptor for assembling a signaling complex. The formation of the signaling complex regulates diverse cellular functions like cell growth, development, and host immune response. Spontaneous activation of RTKs, in the absence of ligands, is often linked to multiple human pathologies like cancers, diabetes, and autoimmune disorders. Researchers are exploring how a cell maintains an autoinhibited state of the enzyme and why such autoinhibition is breached during the progression of human pathology.
Researchers at the Indian Institute of Science Education and Research (IISER), Kolkata, investigated one such RTK called Vascular Endothelial Growth Factor Receptor (VEGFR). The VEGFR family of receptors is the key regulator of the process of generating new blood vessels.
This process is essential for functions like embryonic development, wound healing, tissue regeneration, and tumor formation. Various malignant and non-malignant diseases can be treated by targeting VEGFRs.
The researchers were intrigued by the fact that two members of family VEGFR 1 and VEGFR 2 behaved quite differently. While VEGFR 2, the primary receptor regulating process of formation of new blood vessels, could be spontaneously activated, without its ligand, the other member of the family VEGFR 1 cannot be spontaneously activated even when overexpressed in cells. It camouflages as a dead enzyme VEGFR1 and binds with ten-fold higher affinity to its ligand VEGF-A than VEGFR2. This ligand binding induces a transient kinase (speeding up chemical reactions in the body by an enzyme) activation.
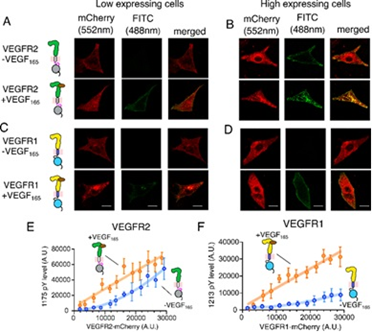
Figure 1: Probing ligand-independent activation of VEGFR. A–D Confocal images of VEGFR2 or VEGFR1 fused to mCherry in low (A, C) and high (b, d) expressing CHO cell lines. The VEGFR expression level is in red, and the phosphorylation status is in green, Scale bar = 10 μm. The expression level of VEGFR2 (panel E) or VEGFR1 (panel F) is plotted against the phosphorylation level of the corresponding tyrosine residues at the C-terminal tail
Activation of VEGFR1 has been found to lead to cancer-associated pain, tumor cell survival in breast cancer, and migration of human colorectal cancer cells.
Probing into why one member of the family is so spontaneously activated and the other autoinhibited, Dr Rahul Das and his team from IISER Kolkata found that a unique ionic latch, present only in VEGFR1, keeps kinase autoinhibited in the basal state. The ionic latch hooks the juxtamembrane segment onto the kinase domain and stabilizes the autoinhibited conformation of VEGFR1.
Exploring the mechanism of the autoinhibited state of VEGFR 1 the researchers proposed a crucial role for cellular tyrosine phosphatase in modulating VEGFR1 activity. The research carried out at the Analytical Biology Facility at IISER Kolkata with its DST-FIST supported ITC and stopped-flow fluorimeter, highlighted the therapeutic potential of phosphatase modulators in regulating VEGFR1-mediated pathological formation of new blood vessels (angiogenesis) which takes place in cancer.
This discovery published in the journal Nature Communication may open new avenues for developing therapeutic interventions against pathological conditions due to the spontaneous activation of VEGFR signaling. The small molecules targeting the autoinhibited state will have a higher potential for treating cancers like human colorectal carcinoma and renal cancer, where VEGFR1 is overexpressed.
Link to the article: https://doi.org/10.1038/s41467-024-45499-2
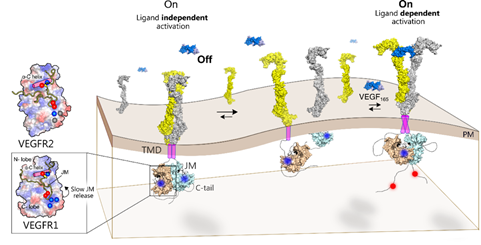
Figure 2: The ligand binding to the extracellular domain (ECD) induces receptor dimerization and rearrangement of the TM-JM segment. Slow release of JM inhibition in VEGFR1 leads to transient tyrosine phosphorylation at the C-terminal tail. Faster release of JM inhibition in VEGFR2 or VEGFR1 mutants remodels the tyrosine phosphorylation to be sustained. Left: Ligand-independent activation of VEGFR1 is suppressed due to a delicate balance between the slow release of JM inhibition and protein tyrosine phosphatase (PTP) activity.
")



")











