








A new study has unravelled mechanisms that lead to certain epilepsies of genetic origin in pediatric population, providing hope for a larger cohort of children suffering from related brain disorders.
Infants with genetic mutations in an intracellular signaling pathway called PI3K-AKT-MTOR pathway often suffer from a range of brain malformations and epilepsy that is mostly non-responsive to conventional anti-seizure medications. Drugs targeting MTOR complexes, such as rapamycin analogues, were observed to be considerably effective to a subset of children harbouring mutations mainly in those complexes. Research in the field has been largely biased towards the direct role of only MTOR (Mammalian Target of Rapamycin) protein behind such disorders, without considering other components of the signalling pathway. There is a distinct lack of focus on a greater proportion of epileptic patients who do not harbour mutations in MTOR. Currently, most of these young patients are left at the mercy of invasive recurrent brain surgeries.
Scientists from JNCASR, an autonomous institute of the Department of Science and Technology (DST), in collaboration with scientists at Seattle Children’s Research Institute, Seattle, USA, focused on other mutations of PI3K-AKT-MTOR pathway and identified acute mechanisms that are MTOR-independent, with the help of preclinical drug assays. The team, led by Dr. Achira Roy, found that acute inhibition of PI3K or AKT, but not MTOR activity, suppresses the intrinsic hyperactivity of the mutant neurons. These acute mechanisms are distinct from those causing neuronal hyperactivity in other AKT/MTOR epileptic models and define parameters to facilitate the development of new molecularly rational therapeutic interventions for different types of intractable epilepsy.
Earlier, the same team had determined that mouse models expressing a patient-related activating mutation in a gene named PIK3CA are epileptic and treatable by inhibition of the respective enzyme. In this study published in the journal Frontiers in Molecular Neuroscience, the researchers recorded brain activity in the same mouse models using different pathway inhibitors and discovered novel physiological mechanisms that are acutely suppressing neuronal excitability.
This research has bridged basic neurobiology and translational biomedical research, that can be the precursor for modern precision medicine. It also highlighted the relevance of repurposing of cancer drugs to treat neurodevelopmental disorders.
This preclinical study provides a foundation to build novel targeted therapeutics towards betterment of livelihood of numerous children suffering from rare brain disorders, including intractable epilepsy.
Publication link: doi: 10.3389/fnmol.2021.772847
For more details, please contact Dr. Achira Roy; Email ID achira[dot]roy[at]jncasr[dot]ac[dot]in.
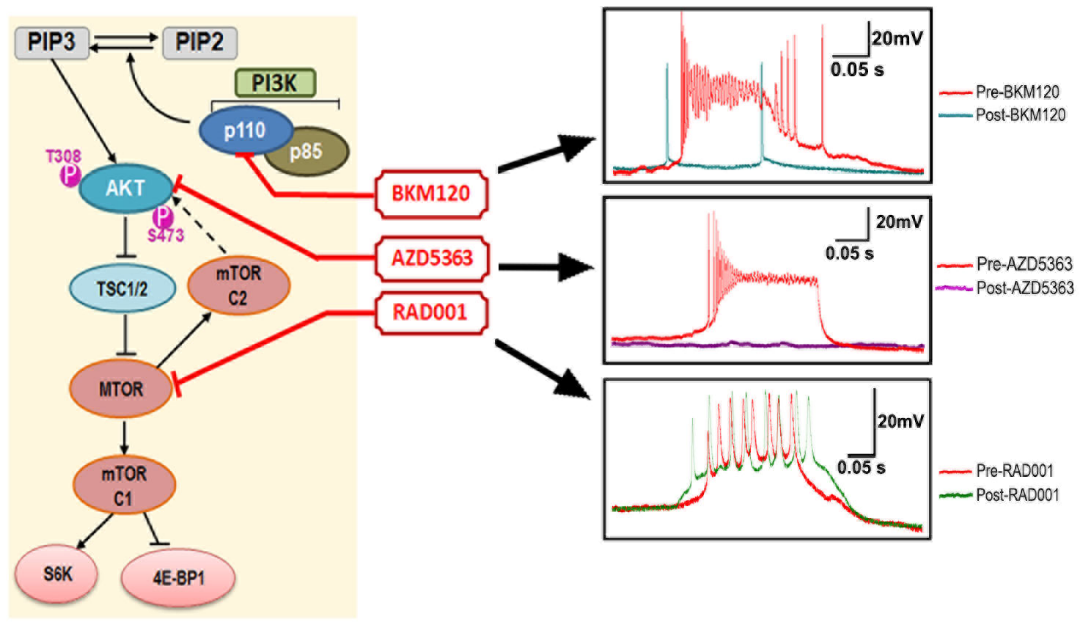
Fig.: Acute inhibition of PI3K (by BKM120) or AKT (by AZD5363), but not MTOR activity (by rapamycin analogue RAD001), preclinically suppresses the intrinsic hyperactivity of the epileptic neurons in Pik3ca mutant mouse brain.
")



")











